

Principal Investigator Training - Role of Principal Investigator in Clinical Research
Launch your rewarding career in clinical research! This blog post explores Principal Investigator training and the path to becoming a leader in medical discovery. Explore PI training options, including accredited online programs, and gain valuable insights into the APIPC™ certification. Invest in your future – enroll in PI training today!

Principal Investigator
Principal Investigator Training - Advanced Principal Investigator Physician Certification (APIPC)™
Are you a physician passionate about advancing medical knowledge through clinical research? Do you aspire to lead clinical trials and contribute to the development of new treatments? If so, then Principal Investigator (PI) training and certification can be your gateway to a rewarding career at the forefront of medical discovery.
What is a Principal Investigator?
A Principal Investigator is a physician who oversees all aspects of a clinical trial. They are responsible for the scientific integrity of the study, ensuring participant safety, and adherence to regulatory guidelines, including those set by ICH-GCP. PIs play a crucial role in ensuring the success of clinical trials and ultimately, the development of life-saving therapies.
Why Consider PI Training and Certification?
While formal PI training is not always mandatory, it equips physicians with the necessary knowledge and skills to excel in this demanding role. Here are some compelling reasons to consider PI training and certification:
Enhanced Knowledge: Training programs delve deeper into the intricacies of clinical trial design, regulatory compliance, Good Clinical Practice (GCP), and ethical considerations.
Increased Credibility: Completing a program and earning an Advanced Principal Investigator Physician Certification (APIPC™) demonstrates your commitment to excellence and expertise in clinical research. This can make you a more attractive candidate for research sponsors and pharmaceutical companies seeking qualified investigators for their trials.
Improved Patient Care: By gaining a thorough understanding of research protocols and ethical considerations, PIs can ensure the well-being of study participants and contribute to the development of safer and more effective treatments.
Career Advancement: As the demand for qualified clinical investigators continues to rise, PI training and certification can open doors to exciting career opportunities in academia, research institutions, and the pharmaceutical industry.
What Does PI Training Cover?
PI training programs typically cover a broad range of topics, including:
Clinical trial design and methodology
Regulatory requirements for clinical research (e.g., FDA regulations, ICH-GCP guidelines)
Ethical considerations in clinical research informed consent, participant protection)
Data management and statistical analysis
Budget development and financial management
Effective communication and collaboration with research sponsors, CROs, and research teams
PI Training Options: Online Convenience with CCRPS
The good news is that you can invest in your PI development through online training programs! The Clinical Research Champions (CCRPS) offers a comprehensive online principal investigator training program designed for physicians seeking advanced review and understanding of essential PI roles. Consider exploring the Medical Monitor Certification as well, which is part of the comprehensive suite of courses offered.
Benefits of CCRPS PI Training:
Accredited Training: CCRPS courses are accredited by the Accreditation Council for Clinical Research & Education (ACCRE) and jointly accredited with PIMED by the American Medical Association (AMA) to provide 17.5 CME credits for physicians.
Enhanced Credibility: The program incorporates ICH GCP and E6 certification, recognized through meeting Transcelerate BioPharma requirements, further bolstering your credentials.
Practical Knowledge: While PI certification through examination isn't mandatory for conducting trials, CCRPS training equips you with the knowledge needed for initial PI roles and paves the way for future certification (which often requires 3,000 hours of PI experience).
Career Advancement: Many physicians utilize CCRPS training to advance their existing PI roles and gain the skills and confidence to function as certified principal investigators. Consider exploring additional certifications such as the Advanced Clinical Research Project Manager Certification to further enhance your career trajectory.
By investing in PI training and potentially pursuing the APIPC™ certification, you're making a strategic move to become a leader in clinical research. CCRPS' online program offers a convenient and accredited way to acquire the knowledge and skills needed to excel in this rewarding field. Take charge of your career and contribute to groundbreaking medical advancements. Enroll in CCRPS PI training today!
The benefits of the PI training course is to improving efficiency, learning about multiple similar trials, and getting the fund of knowledge needed to improve quality, teamwork, and data.
While certification is not required for PIs to conduct trials, PI-sponsors and organization sponsors can show sponsors that PIs receive therapeutic-specific education and content-backed certification.
Most broadly, training can be another quality initiative to prevent trial errors. We teach in-depth with modules covering things like developing inclusion-exclusion criteria, writing protocols, etc. Our focus is on including multiple practical references, applications, and perspectives such that PIs can feel more comfortable making critical decisions in the trial.
Know that non-MDs/PharmDs can be a principal investigator if a physician is a co principal investigator. As a PI, you can take training courses to improve efficiency and apply it through trial examples. The course covers topics such as developing inclusion-exclusion criteria and writing protocols which will prevent errors in future research projects
The benefits of the PI training course is to improving efficiency, learning about multiple similar trials, and getting the fund of knowledge needed to improve quality, teamwork, and data.
While certification is not required for PIs to conduct trials, PI-sponsors and organization sponsors can show sponsors that PIs receive therapeutic-specific education and content-backed certification.
Most broadly, training can be another quality initiative to prevent trial errors. We teach in-depth with modules covering things like developing inclusion-exclusion criteria, writing protocols, etc. Our focus is on including multiple practical references, applications, and perspectives such that PIs can feel more comfortable making critical decisions in the trial.
Know that non-MDs/PharmDs can be a principal investigator if a physician is a co principal investigator.
Principal Investigator Definition
A Principal Investigator in Research is the primary individual responsible for the preparation, conduct, and administration of a research grant, cooperative agreement, training or public service project, contract, or other sponsored project in compliance with applicable laws and regulations and institutional policy governing the conduct of sponsored research.
What Is A Principal Investigator
PI principal investigator is responsible for the preparation, conduct, and completion of the research or project being funded by a grant or sponsor. This is increased by having additional educational training such as through CCRPS’s Advanced Principal Investigator Physician Certification (APIPC)™.
Principal Investigator Salary ranges heavily based on the physicians ability to manage both trials and their typical clinical caseload. While most MDs have a high salary from their career, running a trial can add between $37,000 to $279,000 as their additional clinical research principal investigator salary. Although this can be demanding, the reward of being able to offer new therapeutic advances to their patients is well worth it. Co Principal Investigator is a second physician helping assist in these duties usually for larger trials.
NIH principal investigator funding is best obtained by following and reviewing all of the resources available in the toolkit linked prior. See how to become a principal investigator at the NIH here.
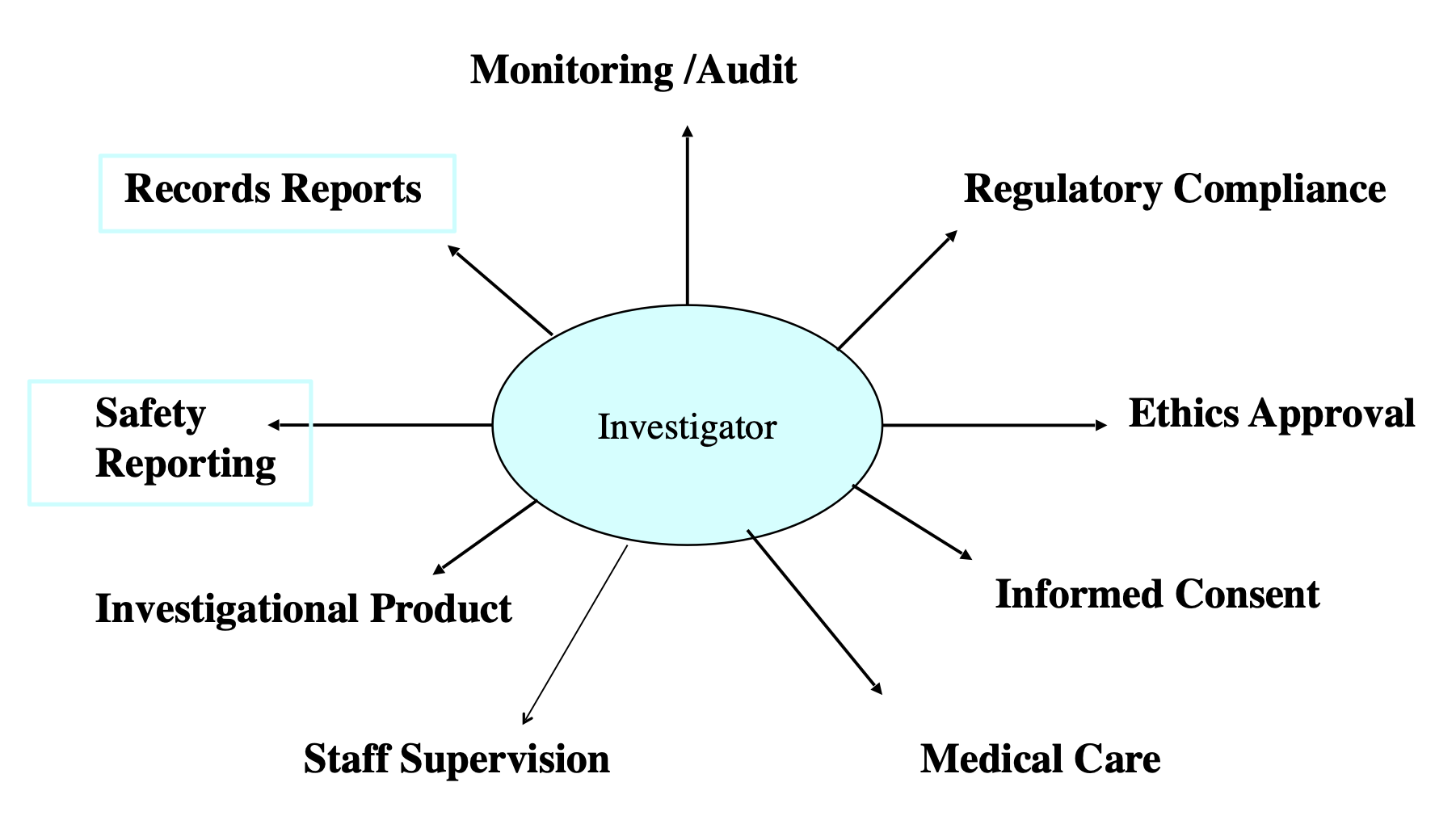
Principal Investigator roles and responsibilities in clinical research
What does a PI do in the clinical research process?
These are covered in the ICH GCP guidelines and are summarized below (Feehan, 2020):
Role of Principal Investigator in Clinical Research: Anyone qualified by training to run the trial; a physician or dentist must be listed as a sub-investigator if the principal investigator is not a physician
TO: Hire and train qualified individuals to run the trial AND
Protect subject safety: Protect subjects from harm, Keep track of drugs and distribute only as specified in the protocol, Obtain informed consent, Ensure IRB approval, Keep careful records and maintain them for as long as the protocol dictates or at least 2 years AND
Report: Progress, safety, financial, and a final report to the study sponsor, Adverse events; serious adverse events must be reported immediately, Update financial disclosures if any circumstances change during the study AND
Follow Form 1572: Strictly adhere to the protocol, Directly supervise the study and take responsibility for study staff, Inform subjects of experimental nature of the drug products, Report adverse events and stay updated on the investigational brochure, Maintain records, Ensure IRB compliance AND
Prepare for FDA inspections: Ensure all records are complete and easily accessible by FDA, Send a written response within 15 business days if any violations are found AND
Avoid violations: Read all communications from the IRB, Hire experienced staff and verify their credentials, Train staff regularly, Check for conflicts of interest/financial disclosures regularly, Write efficient protocols or reduce inefficiencies or confusing portions of the protocol, Keep regulatory binders up to date and conduct continuing reviews, Meet with the team regularly, Conduct several dry runs to ensure the study will run smoothly, Regularly check data processes
Download the FDA Principal Investigator roles and responsibilities powerpoint here.
Principal Investigator Training
The responsibility of conducting successful clinical trial lies upon the shoulders of a Principal Investigator. This successful completion of the trials is accomplished by major duties of an Investigator who is responsible for the delegation of duties to other staff members as well as the safety, protection of rights and well-being of the volunteers.
The research volunteers have the right to know about the study that either it is conducted for research purpose or not. Moreover, any alterations in study protocol must be appropriately mentioned to the volunteers and giving them the right to either continue or quit the studies. Providing accurate and appropriate data, informing the ethical committees about the issues related to research site, issues related to adverse effects of drug are the key responsibilities of the Investigator.
To perform all these crucial responsibilities an investigator must pass through the training and evaluation to improve the standards of trials.
The training of PI’s is categorized into different modules so that with the passage of each module all the responsibilities and documents become clear.
PI Course Syllabus
Introduction
Accreditation Statement
CME Handout - How to Obtain 17.5 CME Credits through AMA/ACCME
Principal Investigator Toolkit
How to Effectively Use this Course
The Role Of The Principal Investigator
Principal Investigators Roles, Checklists, & GCP Guidelines
Principal Investigators Reporting Responsibilities for AEs and SAEs
FDA Form 1572 - Part 1
FDA Form 1572 - Part 2
Investigator Initiated Multi-Center Trials
Investigational Product Storage and Dispensing
Investigational Product Accountability in Clinical Trials
Clinical Trial Design & Protocol
Phases of Clinical Trials
Designs of Clinical Trials
Randomized Controlled Trials
Institutional Review Board (IRB)
The Clinical Trial Protocol - Advanced Mastery Review
Protocol Deviations and Violations
Inclusion and Exclusion Criteria in Clinical Research
IND Application
IND and NDA Process
Documents & Informed Consent
Source Documents and Informed Consent Forms
Informed Consent (ICH GCP Section 4.8)
Trial Management, Data Handling, and Record Keeping
Compliance with E-Signatures CFR 21 Part 11
Essential Regulatory Documents Guidance and Binder Tabs (Part 1)
Essential Regulatory Documents Guidance and Binder Tabs (Part 2)
Guidelines for Designing and Completing Case Report Forms
Do’s and Don’ts of a Case Report Form Design
Investigators Brochures
Trial Master File and DIA Model
Trial Master File Reference Guide
Financial Disclosure- Duties and Strategies for Clinical Studies
Financial Disclosures and Conflicts of Interest in Clinical Research
Adverse Events
Advanced Review of Adverse Events
Reporting of Adverse Events
Safety Reporting Requirements for Sponsor Investigators of An IND
Site Visits And Audits
Overview of Types of Monitoring Visits
Site and Investigator Selection
Site Selection/Qualification Visit (Pre-Study Visit)
Site Close Out Visit
Audits vs. Inspections
FDA Warning Letter
Site FDA Audit Inspection Checklist
How to Survive Through an FDA Inspection
Do and Don’ts during an FDA Inspection
Patient Safety, Recruitment, And Compliance
Introduction & History of ICH GCP
Compliance Requirements in Clinical Trials
Subject Recruitment and Retention (Part 1)
Subject Recruitment and Retention (Part 2)
Safety of Human Subjects in Clinical Research
Ethics of Research Involving Pregnant Women and Fetuses
Ethics of Research Involving Mentally Incapacitated
Ethics of Research Involving Children
Scientific Misconduct in Research and How to Prevent It
Increasing Subject Compliance in Clinical Trials
Misconduct in Research – Detecting Falsification
Self-Assessments
Self-Assessment MiniQuiz 1
Self-Assessment MiniQuiz 2
Self-Assessment Quiz A
Self-Assessment Quiz B
Final Quiz
The Drug Development Process for PIs:
In the first module of training, PI is introduced with the clinical research trials, phases, pre- clinical research, drug developing method and dealing with common difficulties. PI must be aware of the purpose of clinical trials which are conducted to develop a drug to ultimately leading to the capable the drug used for prevention, diagnosis and treatment of disease.
The main objectives of the clinical trials include:
Treatment is the common fascination for the subjects to participate in the trials as
it provides them with medicine for treatment and diagnosis.
Observational studies enable the investigator to find the relationship between the
habits of socioeconomic group and the progress pattern of disease.
Prevention type of studies include subjects who want to prevent the disease rather
than treatment. Such kind of studies require the personnel having family history of
the disease.
Diagnostic type of trials is used to treat the disease efficiently.
Basic science by its nature allows to concentrate upon the phenomenon, closed
observation, experimentation of hypothesis, extracting conclusion to develop a new approach.
Pre-clinical trials as the name indicates are performed before the clinical trials and commonly known as Phase 0 trials involve First-in-man trials to know about the safety of the drugs and allows the determination of amount of safe dose of drug. This kind of trial is used to test the new medical devices, diagnostic tools, gene therapy and drugs.
There are mainly 4 phases of clinical research trials.
Phase 01. Determination of pharmacology and tolerability
Phase 02. Evaluation of safety and efficacy
Phase 03. Evaluation of safety and risk ratio as well as effectiveness. Successful completion of phase 3 trials leads to submission of application for the FDA approval.
Phase 04. Monitoring long term effects and effectiveness. Successful phase 4 trials are considered as FDA approved drug.
The Role of Principal Investigator:
A person responsible for the conduct, preparation, research grants, agreements, training or public service project, contract or other sponsored projects in compliance with applicable laws and regulations and policy governing the conduct of sponsored research.
FDA recommends a form duly filled and signed by the investigator which serves as a statement from the side of investigator is known as FDA form 1572
Major Roles of PI:
The major roles of the PI are The Position roles (managing the integrity of design and collaborative results, reporting to the individuals like dean , department head and divisional chief),coordination with the administration, scientific proposal preparation, arranging and managing budgets, preparation of protocol according to the regulations and assuring the approval of any changes before implementation, acceptance of award in the light of rules and regulations, Conducting research (supervision to implement the rules, supervision of the team to ensure ethical conduct, reporting any misconduct), applying appropriate cost rate to the facilities and administrative, allocation of space to conduct the research, arranging equipment for study etc.
ICH has provided adequate GCP for Investigator to work effectively. These guidelines provide information about following aspects:
Qualification and dealing with the agreements.
Adequate resources in terms of staff and budget.
Protection of the rights and safety of trials subjects either human or animals.
Procedure of communication with regulatory bodies like IRB/IEC.
Following and implementation of the rules and protocols set by regulatory bodies,
Allowing the supervision of the investigational product to a qualified and responsible
person.
Responsibilities regarding design of studies (randomization and blinding).
Arranging the informed consent of individuals.
Keeping records and reporting to the regulatory bodies.
Safety reporting
Termination or suspension of the trials before time.
Submission of final reports.
Informed Consent:
A document signed by every volunteer participating in the trial by informing him /her regarding all the matters of trials like, either he /she is voluntarily participating in the trials, informed about the risks, clarifying the role in trials and procedure of study. Federally, it is compulsory to get signed informed consent before participation in the clinical trials.
This link provides all the information regarding inform consent form and ICF review sample.
To effectively complete the consent process, interviewer should provide sufficient time to the participant to think that either he should participate or not. This process also allows the participants to review the procedure and frequently ask any query related to the trials. This process must be transparent and free of influences.
For developing a consent form standard UCI, IRB consent template must be followed. It must include the following key points.
General information regarding purpose, design, and statement of study.
Objective of the study must be clear including an appropriate definition that why
FDA testing is necessary.
Complete description of the process either in tabular or descriptive form comprising
of complete description of stages involved in process, inclusion/ exclusion criteria,
type of questions in case of questionnaire.
Risks associated with the study must be properly defined in tabular or descriptive
form. If risks are not clear, then a statement must be added that any unforeseeable
risk can occur.
To clarify the participant can withdraw or Investigator can terminate any participant
at any time. It is investigator’s responsibility to clarify the procedure of the
withdrawal from the study when participant wants.
Confidentiality must be maintained regarding the subject’s information, procedure,
and research data and about the record keeper.
Investigator will provide new information to the subject by standard text.
Or the benefit of the participant who wants to withdraw from the studies, extra
courses or benefits must be explained in an appropriate way.
Research member’s financial interest in the study must be mentioned as well as the
specimen collection should be informed.
It must be clearly written that the subject has the right to either participate or not in
the study.
Signature line for participant and authorizing member must be included. Witness
line shall also be included in some kind of studies where necessary.
The Clinical Research Protocol:
Protocol is the document describing the procedure of conducting clinical trials ensuring the safety of subjects or volunteers and integrity of collected data. This document must comprise of following headings and details regarding trials:
Title page
Background information
Objective /purpose
Study design
Inclusion and exclusion of subjects
Treatment of subjects
Evaluation and assessment of safety and efficacy
Adverse events
Suspension of study
Statistical data
QC and QA
Ethics
Data handling and record keeping
Publication policy
Project timetable
References
Appendices
The NIH also provides templates and resources to develop the protocols and designs to conduct the trials.
When the activities without any significant reason diverge from the IRB approved protocol the is known as protocol deviation.
Adherence to the trial related requirements, following GCP requirements, and fulfilling regulatory requirements is known as Protocol compliance.
Understanding Adverse events:
Adverse event is defined as an unintended and unwanted sign, symptom or disease partially associated with the use of drug without any judgement about causality and relationship to the drug.
If a sponsor or investigator based on outcomes (death, life threatening adverse reaction, In-patient hospitalization or disruption life functions) considers the adverse event or reaction serious.
All adverse events should be documented in the patient’s medical record. To collect AE’s patients should not be asked anticipated questions either they should be asked the open- ended questions, during examination and evaluation. A progress note must contain good clinical practice and good clinical practice research. Report must contain date of AE initiation, attribution of AE, date of resolved AE, documentation of worsen or untreated AE.
Recording of AE onto a case report form (CRF) includes details:
Date of initiation of AE
Treatment of AE
Garde of severity
Attribute of AE
AE resolved date.
For worsen condition or treatment changing progress or any relationship should be
documented on the other CRF form
Reporting to regulatory bodies involves routine and expedited.
IND Safety reports (ISR) comprised of all types of adverse events, In vitro testing details, findings of other studies and all other suspected adverse events.
Format for submission of an ISR include Narrative ( all kind of data either published or unpublished analysis data),
FDA form
Council of International Organization Medical Sciences Form
Source Documentation
A pharmaceutical company or sponsor provide or sends the new drug for approval to the FDA including all the data of clinical trials. The FDA then asks for the provision of the source data from where it was captured. This documented data is called source document.
Essential documents in clinical trials
Investigator’s Brochure, study protocol, Informed consent or subject information, reports of research trials and case report form.
Firstly, Investigator brochure (IB) contains information about the investigational drug before and after the performing he clinical studies in a brief and concise manner. This document is comprised of keywords and abbreviations, contents list, brief description of the investigational drug, general approach towards study and brief description as introduction, characteristics of the medicinal product, nonclinical and clinical studies and at the end conclusion and references.
Secondly, Clinical Study Protocol is a documentation of goals, objective, and design of clinical study. This document is designed after the instructions of all parties participating in clinical trials and this document should contain all the information regarding clinical trials and then sent to the authorities for review.
Thirdly, when any amendment is made in the study protocol, protocol amendment document is used. Before implementation it must be again approved by the authorities.
Fourth, Informed consent is the document to ensure that the volunteers are perfectly aware of the objective of trials, Investigational product. This document depends upon the willingness of the volunteers and they have right to leave the trials.
Fifth, Study progress reports are prepared by the medical monitor either on daily basis or final report is prepared to show the committees.
Sixth, Case record form is the document to record the data of individuals involved in the study. It could either be an electronic document.
Responsibilities of an investigator according to GCP guidelines include:
Supervision of the conduct of clinical investigation.
Delegation of the duties to the qualified personnel.
Training of the participating staff
Arranging an individual for the supervision of each site.
Protection of rights, safety and welfare of study objects.
Communication with IRB requires high level of professionalism for which PI assigns this duty to Regulatory document specialist (RSD) and documentation of this communication is maintained.
SOP’s for communication are provided in this link.
Deviation’s fraud and noncompliance:
When IRB or any committee sets regulations, rules, policies, and laws and rely upon the organization or any person to imply them, then any failure in implementation of these rules is commonly described as noncompliance. To overcome this issue, NLM has created a website and provided the time frame to government and private trials conducting bodies to register and provide the results within the given time frame.
A term fraud is commonly used in clinical trials which means contravention of faith and dealing intentionally, to harm any individual by manipulating the research data and results. It also includes deviating from the set protocols, policies and manipulating data and research results. The question arises that how and why fraud occurs in clinical trials. There are some points summarized to explain it.
To gain fame by participating in internationally renowned trials.
Manipulating data of repeated testing due to lethargy of research staff or
investigator.
Rule’s policies and incentives attached to the trials are the environmental factors.
Idiosyncrasies, ego, competition among colleague investigators, for increasing the
tenure and promotion.
When an investigator either intentionally or unintentionally fails to comply with the policies and regulatory requirements, FDA has authority to disqualify him either permanently or temporarily. This disqualified investigator is not allowed to conduct any kind of investigations regulated by FDA.
The main steps or headings of disqualification of an investigator by FDA are described here.
• The Disqualification processes.
o Issuance of notification for disqualifying an investigator and providing an opportunity to explain.
o Consent agreement
o Summon of hearing as an opportunity after disqualification. o After hearing taking crucial decisions
o Actions upon disqualification:
Criminal prosecutions
Revealing the information regarding decision of disqualification
Reinstatement of disqualified investigator
The link for the detail study: FDA proceeding